
技术问答
代谢组学数据处理——数据归一化
1月 30, 2019
多维统计模型建立之前,首先需要对数据作归一化(Normalization)处理(有些学者称为标准化Standardization),一是让数据无量纲化,使不同性质的变量具有可比性;二是将不同数量级的变量数据经过不同的转换(transform)至合适范围,避免大值变量掩盖小值变量的波动。在代谢组学数据处理中,常用的归一化方法有Ctr(Center scaling), UV (unit variance scaling)和Par(Pareto scaling)。Ctr也叫中心化是原数据减去每列变量的均值,UV是数据中心化后除以列变量标准差(Standard deviation),Par是数据中心化后除以列变量标准差的算术平方根。Ctr将原数据转化成离原点更近的新数据,可调节代谢物的高低浓度差异;UV的优势是所有变量拥有同等的重要性,但缺点是检测误差可能会被放大;Par相比于UV更接近于原始测量数据,但缺点是对变化倍数大的变量更敏感[1]。UV和Par是常用的归一化方式,基于不同的归一化方式后续的数据分析将选择不同的差异代谢物筛选方法,如UV下常使用V-plot(图1-A),Par下则常用S-plot(图1-B)。无论选择何种归一化方式,都需要对建立的模型作严格验证以确保筛选出可靠的差异代谢物。因为VIP值通常用于差异变量筛选标准之一,V-plot可比较客观的选择出变量。对于Biomarker Discovery的诊断工具,我们推荐使用V-plot和相关性Corr.Coeffs. 的p值同时考虑的标准,如下图2所示。
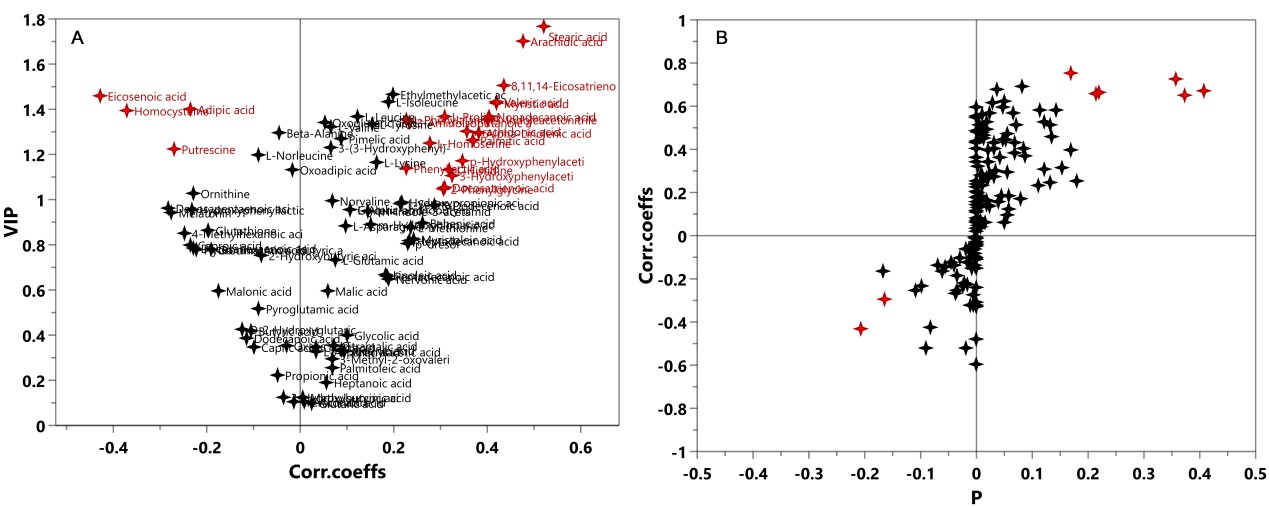
图1. V-plot和S-plot示意图
图2. 基于VIP和Corr.Coeffs的p值的V-plot用于差异代谢物的筛选。来源:麦特绘谱XploreMET软件。
