
文献解读
客户案例 | 肠肝联动新发现:NAD+信号如何‘串通’ILC2s,影响溃疡性结肠炎
炎症性肠病(IBD),包括溃疡性结肠炎(UC)和克罗恩病,是临床上主要的慢性和复发性肠道炎症疾病。在其他器官(如肝脏)存在异常的IBD患者中,往往存在IBD的严重程度增加,然而,肝脏疾病加重IBD的机制尚未完全了解。在哺乳动物中,肝脏通过色氨酸从头合成烟酰胺腺嘌呤二核苷酸(NAD+),并将烟酰胺(NAM)释放到血液中,以供外周组织通过NAD+补救途径来满足NAD+的需求。越来越多的证据表明,NAD+代谢在编程肠道结肠炎期间的免疫反应中起作用,但尚不清楚主要循环自肝脏的NAD+是否通过调节肠道免疫细胞来影响结肠炎的组织修复。
近期,山东大学齐鲁医院消化内科李石洋教授、左秀丽教授和基础医学院袁得天教授在Advanced Science发表了题为Sensing of liver-derived nicotinamide by intestinal group 2 innate lymphoid cells links liver cirrhosis and ulcerative colitis susceptibility的研究文章,通过代谢组学、单细胞转录组学、基因编辑和骨髓移植等技术手段,揭示了慢性肝病扰乱肝脏NAD+代谢,导致经肝-肠轴到达肠道的NAD+合成前体物NAM减少,损害肠道2型固有淋巴样细胞(ILC2s)的组织修复功能,继而在并发UC时加重疾病。(麦特绘谱提供13C6-Glucose代谢流检测服务)

研究思路
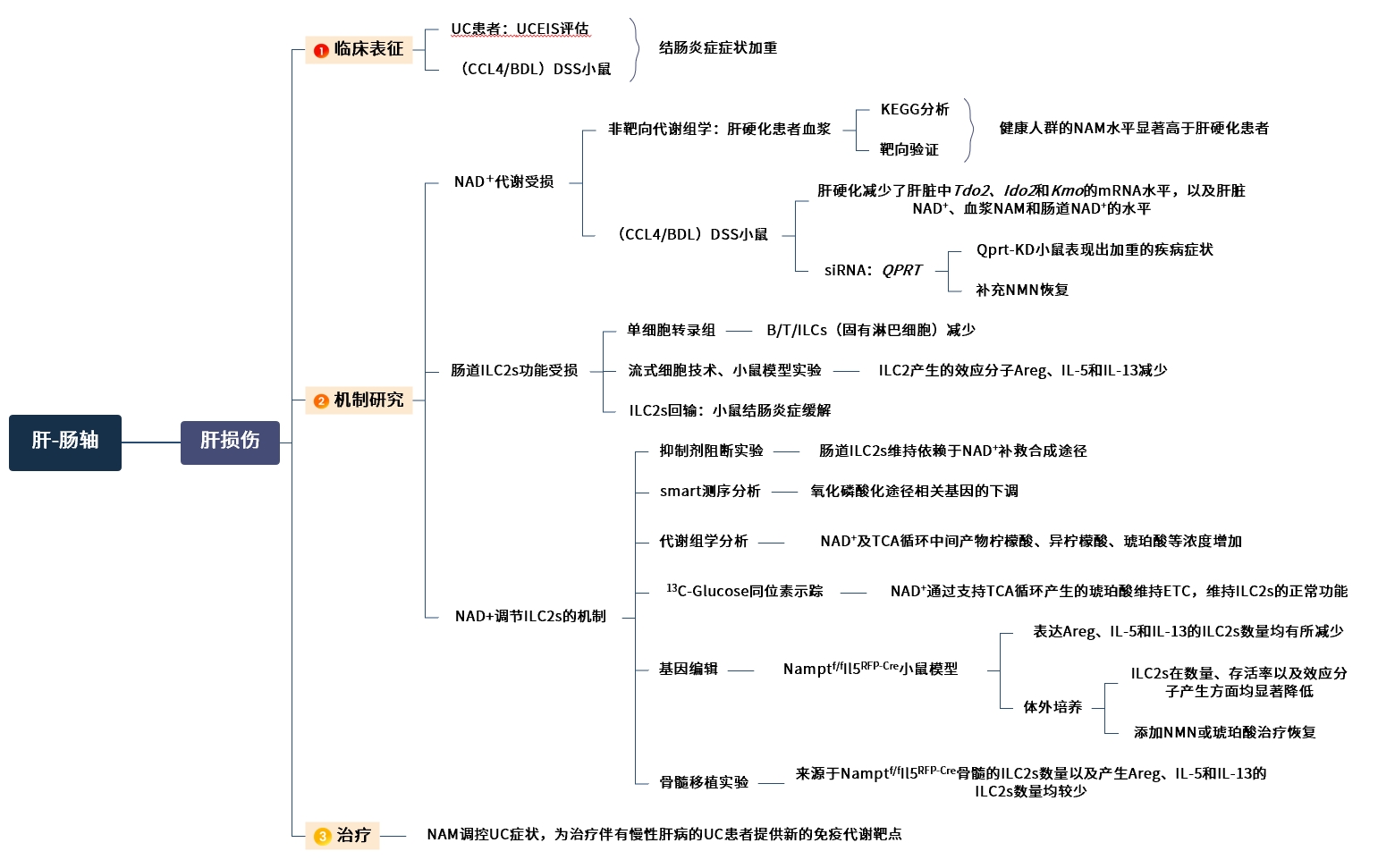
图1. 技术路线
研究结论
1. 肝损伤和溃疡性结肠炎的症状相关
首先研究了肝损伤对结肠炎的影响,采用回顾性图表分析了UC患者血清中丙氨酸转氨酶(ALT)和天冬氨酸转氨酶(AST)水平分类的溃疡性结肠炎内镜严重程度指数(UCEIS),发现肝损伤加重了UC患者的结肠炎症状,两者呈现正相关关系,该结论在四氯化碳(CCL4)诱导和胆管结扎(BDL)肝损伤结合DSS实验性结肠炎小鼠模型中得到验证。

图2. 肝损伤与UC和实验性结肠炎的严重症状相关
2. 肝损伤损害人和小鼠NAD+代谢
进一步探索肝损伤加重结肠炎相关的机制,采用非靶向代谢组学对健康供体和肝硬化患者的血浆进行研究,发现有67种代谢物在肝硬化患者中减少,KEGG分析发现辅助因子的生物合成途径变化最大,这是由L-丝氨酸、烟酰胺(NAM)、泛酸和视黄醇水平降低,以及脱氧胆酸3-葡糖醛酸和L-酪氨酸水平的增加造成的。对肝硬化患者血浆NAM进行检测,发现健康人群的NAM水平显著高于肝硬化患者。
RNA-seq数据显示非酒精性脂肪性肝炎和酒精性肝炎患者肝脏样本中,参与NAD+从头合成和补救合成途径的关键酶(TDO2、HAAO、QPRT)mRNA水平降低。在肝损伤小鼠模型中进行验证,发现肝脏中Tdo2、Ido2和Kmo的mRNA水平,以及肝脏NAD+、血浆NAM和肠道NAD+的水平均有所降低。
使用AAV2/8载体携带针对QPRT的mir30-shRNA(Qprt-KD),特异性敲低肝脏中的QPRT基因,成功降低了肝脏NAD+、血浆NAM和肠道NAD+的水平,在DSS诱导的结肠炎模型中,Qprt-KD小鼠表现出加重的疾病症状,而补充NMN(烟酰胺单核苷酸,NAD+前体)能够逆转这一趋势,表明肝脏NAD+代谢对结肠炎进展有重要影响。
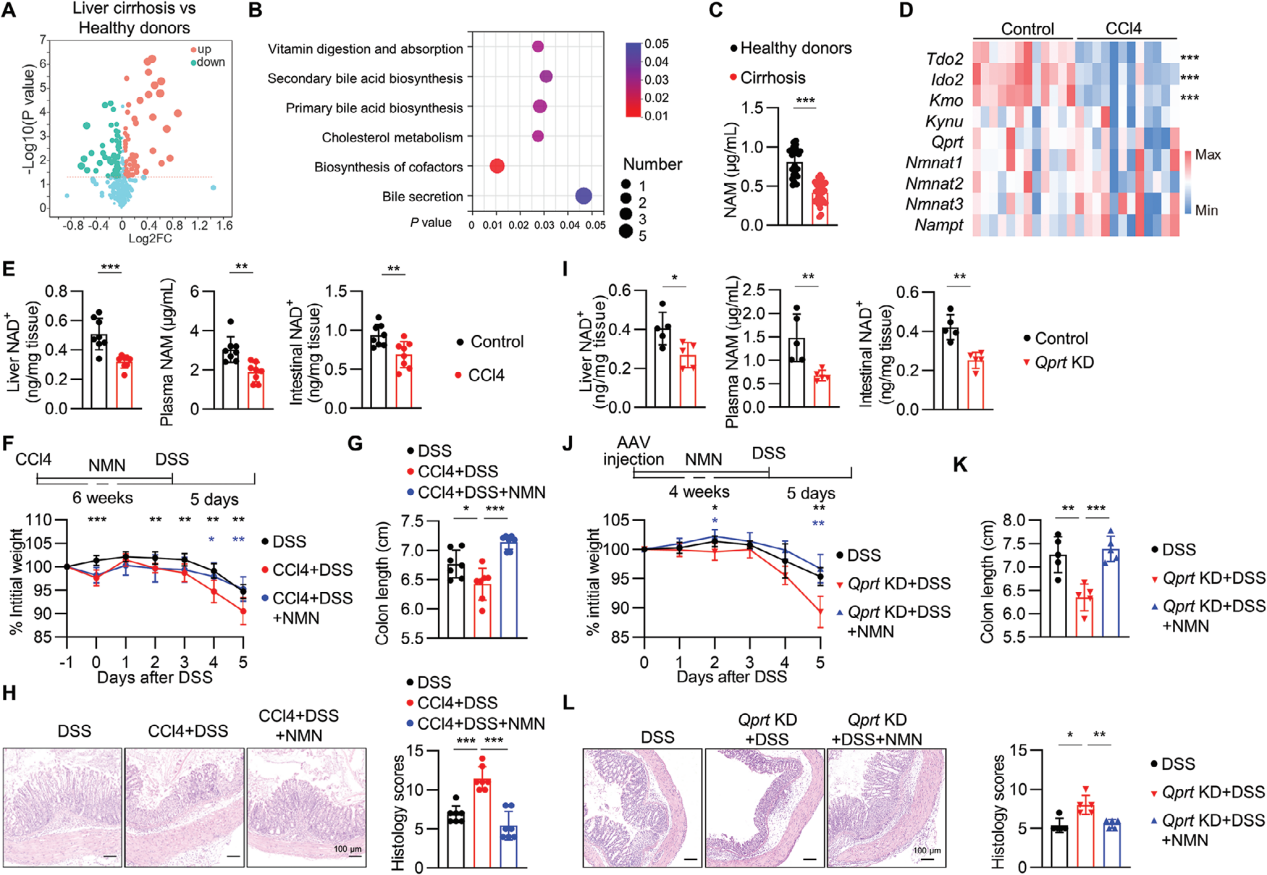
图3. 肝损伤损害人和小鼠NAD+代谢
3. 肝损伤破坏肠道ILC2s
在分子层面继续探索肝损伤加剧结肠炎的机制,采用单细胞转录组学对CCL4处理组小鼠进行研究,发现B/T/ILCs(固有淋巴细胞)减少。通过流式细胞术分析和小鼠模型实验,确认CCl4处理减少了肠道ILC2s产生的效应分子Areg、IL-5和IL-13。将纯化的肠道ILC2s回输到CCl4处理的小鼠中,随后进行DSS处理,发现恢复ILC2s功能能够缓解CCl4加剧的DSS结肠炎。
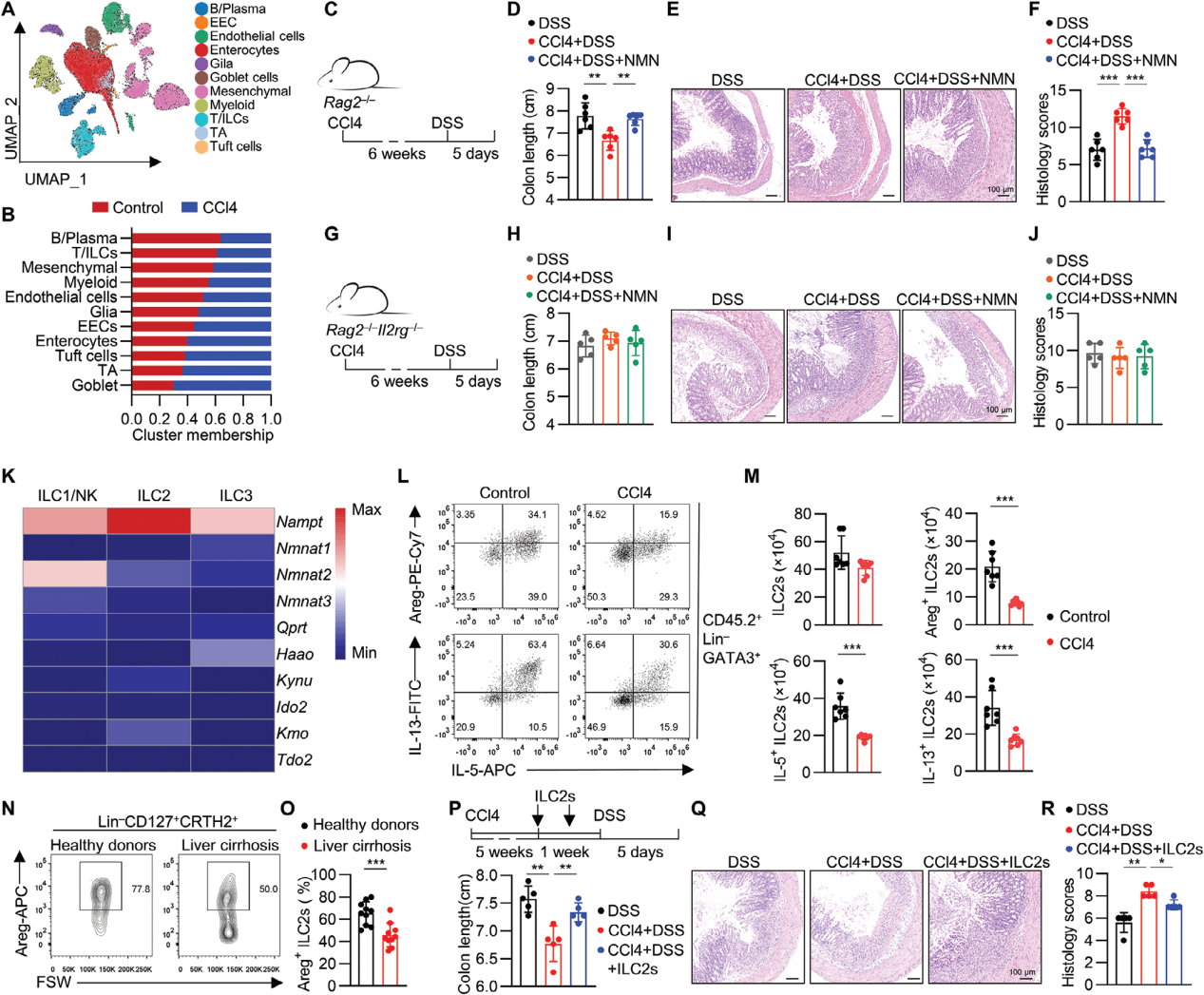
图4. 肝损伤破坏肠道ILC2s
4. 肠道ILC2s功能维持依赖于NAD+补救合成途径
为了评估NAD+代谢是否直接调控ILC2s,使用不同的抑制剂来阻断NAD+合成的不同步骤,GTN(NMNAT的抑制剂)显著抑制了ILC2s的细胞活性和效应分子的产生,表明NAD+的生成对ILC2s的功能至关重要;FK866(NAMPT的抑制剂)显著降低了ILC2s的存活率和细胞因子产生,进一步证明了NAD+补救合成途径对ILC2s功能维持的重要性。
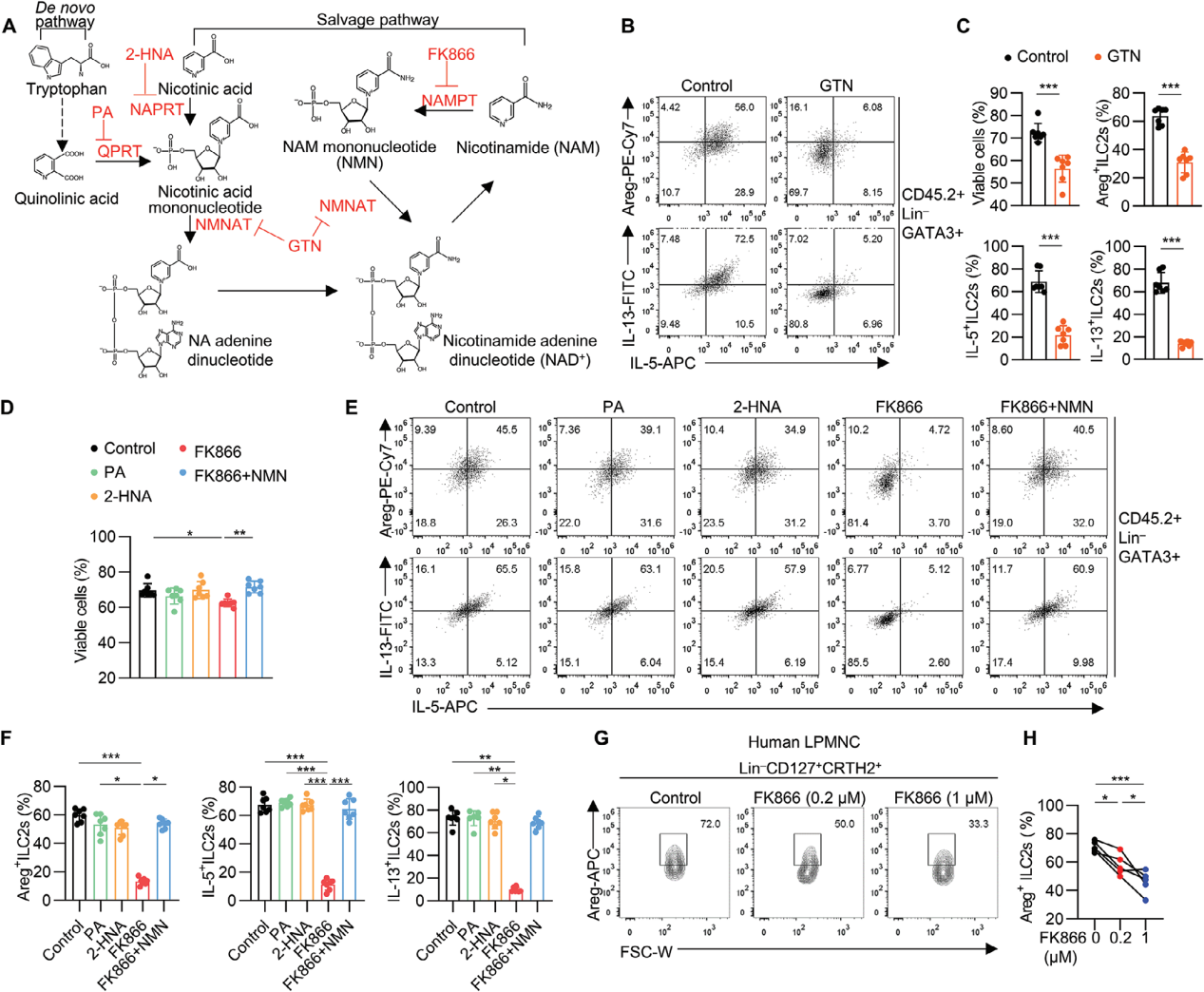
图5. 肠道ILC2s维持依赖于NAD+补救合成途径
5. NAD+调节ILC2s的机制
NAD+作为电子受体,在糖酵解和三羧酸循环(TCA)中接受高能电子,最终将这些电子传递给电子传递链(ETC)的复合体I,驱动氧化磷酸化过程,这是细胞产生三磷酸腺苷的主要途径。
ILC2s的SMART测序转录谱显示NAD+合成抑制剂FK866处理后,大量基因表达发生变化,特别是细胞周期和氧化磷酸化途径相关基因的下调。进一步的代谢组学分析显示,IL-33激活ILC2s时,NAD+及TCA循环中间产物柠檬酸、异柠檬酸、琥珀酸等浓度增加,表明NAD+对TCA循环和氧化磷酸化的促进作用。
使用13C标记的葡萄糖追踪ILC2s探究NAD+是否通过TCA循环代谢产物调控ILC2s,结果显示FK866处理导致ILC2s中丙酮酸和苹果酸积累,减少了琥珀酸及其13C标记的同位素体,以及NAD+和NADH的含量。补充TCA循环中间产物对NAD+和NADH水平无显著影响,但琥珀酸能够部分恢复FK866抑制的ILC2s功能,表明NAD+可以通过支持琥珀酸生成来维持ILC2s。
使用丙二酸单乙酯钾盐(EM-K+)阻断琥珀酸向ETC的电子传递时,琥珀酸对FK866处理的ILC2s恢复作用被消除,进一步论证了NAD+通过支持TCA循环产生的琥珀酸维持ETC,从而保持ILC2s的正常功能。
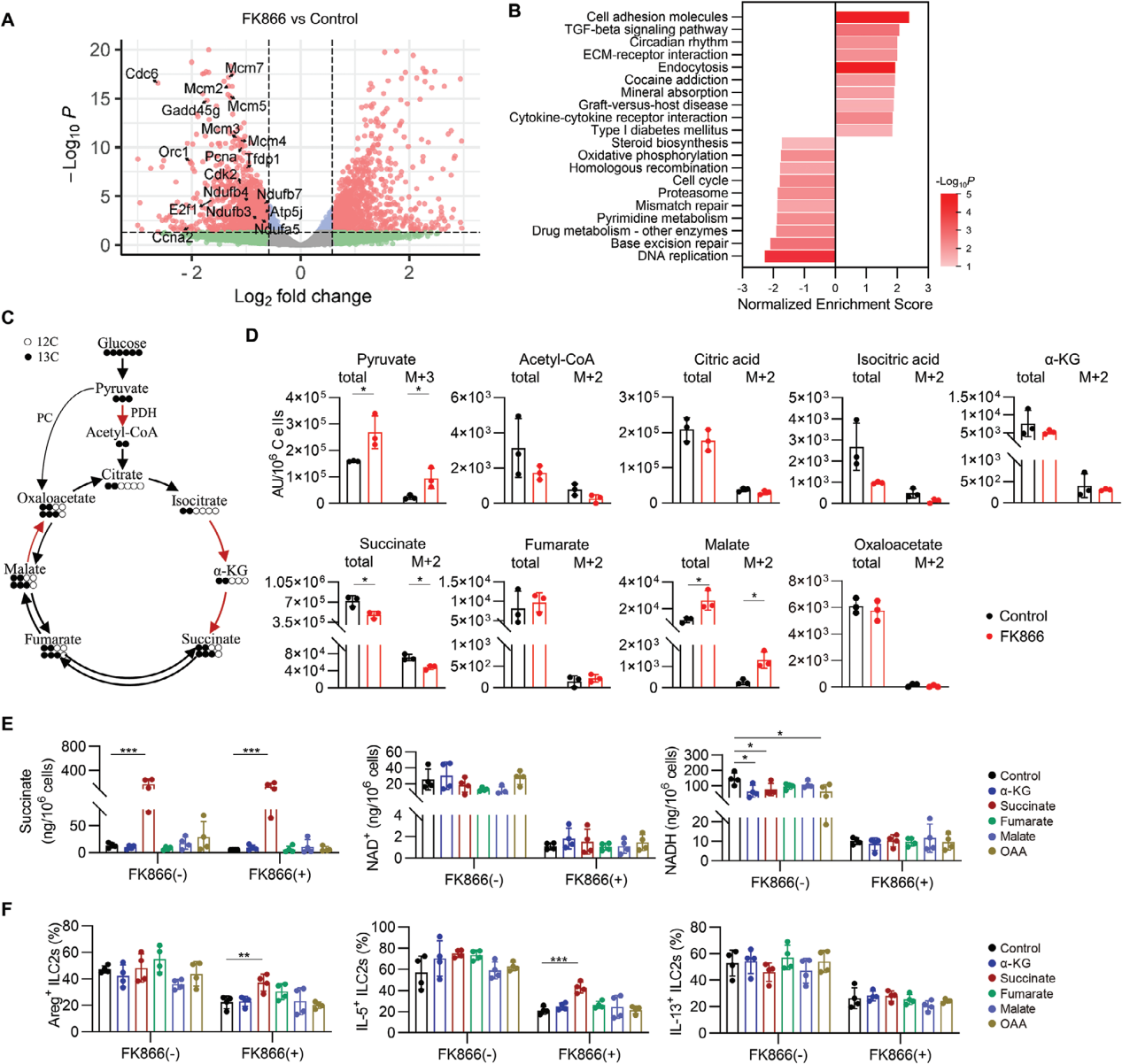
图6. NAD+通过支持琥珀酸盐维持肠道ILC2s
6. NAD+以细胞内在方式调节肠道ILC2s
从遗传学的角度进一步探究NAD+对ILC2s的调控作用,利用基因编辑技术构建Namptf/fIl5RFP-Cre小鼠模型,发现表达Areg、IL-5和IL-13的ILC2s数量均有所减少。从Namptf/fIl5RFP-Cre小鼠中分离出ILC2s,并在体外培养系统中加入IL-2、IL-7、IL-25和IL-33等细胞因子,与野生型对照小鼠相比,Namptf/fIl5RFP-Cre小鼠来源的ILC2s在数量、存活率以及效应分子产生方面均显著降低。通过添加NMN或琥珀酸治疗,可以恢复这些ILC2s的存活率和功能,支持了NAD+通过琥珀酸向电子传递链供电子以维持肠道ILC2s的存活和功能的机制。
通过混合骨髓移植实验进一步证实了NAMPT在ILC2s中的内在作用,来自Nampt+/+Il5RFP-Cre(CD45.1/CD45.2)和Namptf/fIl5RFP-Cre(CD45.2/CD45.2)小鼠的骨髓细胞以1:1的比例混合,并转移到Rag2–/–Il2rg–/–小鼠中。六周后,从嵌合小鼠的肠道中分离出ILC2s,并通过同系标记追踪其来源。结果显示,来源于Namptf/fIl5RFP-Cre骨髓的ILC2s数量以及产生Areg、IL-5和IL-13的ILC2s数量均较少。
给Namptf/fIl5RFP-Cre小鼠和同窝对照组喂食DSS, Namptf/fIl5RFP-Cre小鼠结肠炎更严重,结肠缩短,组织学严重程度增加,添加NMN后症状减轻,此外,NMN也使DSS处理的Namptf/fIl5RFP-Cre小鼠ILC2s数量以及Areg、IL5和IL-13得到恢复。
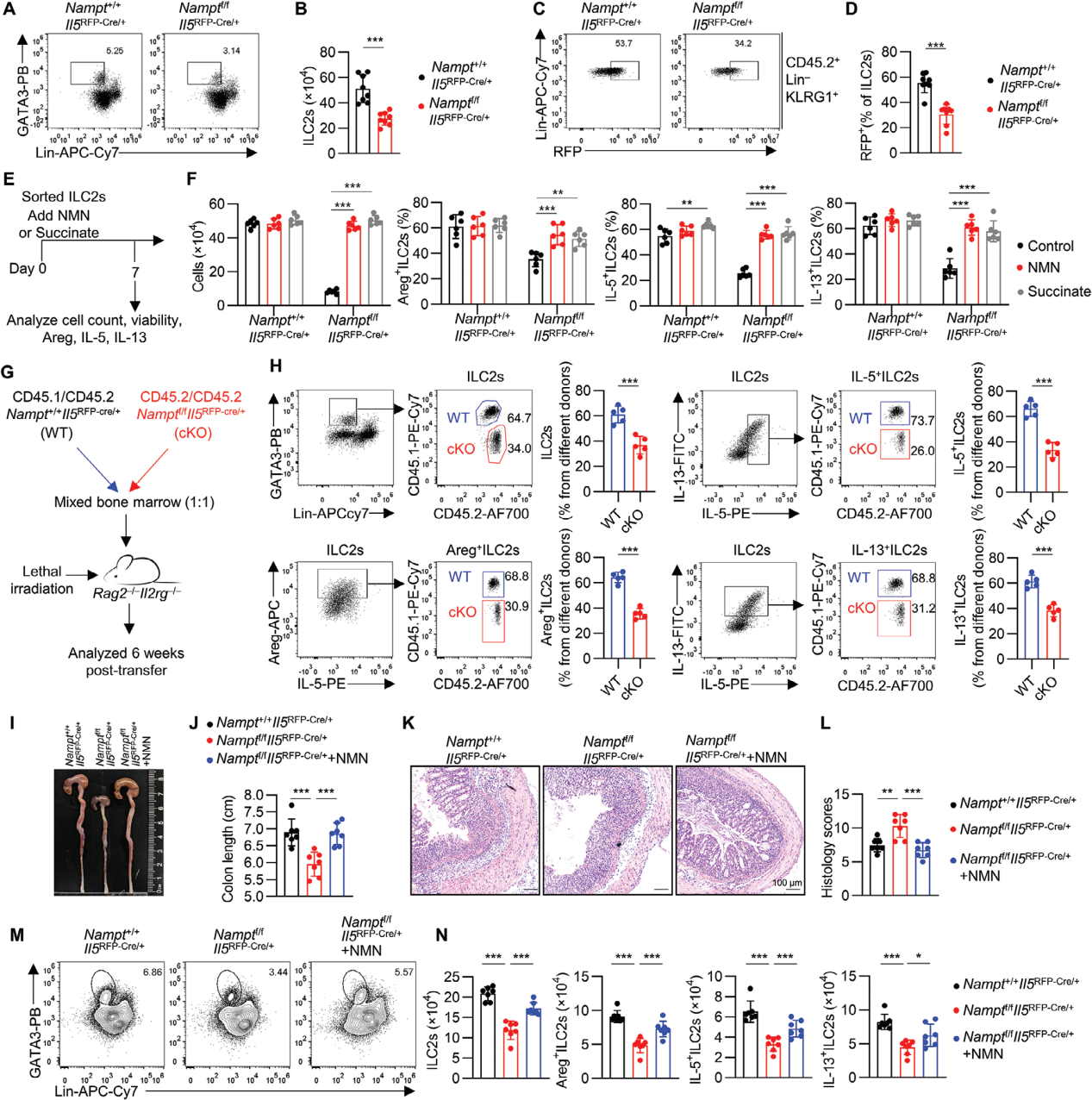
图7. ILC2s 中NAMPT缺乏可加重DSS诱导的结肠炎
小结
本研究揭示了慢性肝病加重UC的免疫代谢调控机制,发现肠道ILC2s通过感知肝脏来源的NAM来调控UC症状,为治疗伴有慢性肝病的UC患者提供新的免疫代谢靶点。
参考文献
Sensing of Liver-Derived Nicotinamide by Intestinal Group 2 Innate Lymphoid Cells Links Liver Cirrhosis and Ulcerative Colitis Susceptibility. Advanced Science. 2024
请扫描二维码阅读原文

绘谱帮你测
本研究使用麦特绘谱提供的13C标记的葡萄糖代谢流检测服务,发现NAD+可以通过支持琥珀酸生成来维持ILC2s的正常功能,揭示了NAD+调控ILC2s的功能机制。麦特绘谱提供全面的代谢流检测服务,可追踪含13C和15N等被标记物100+种,全面覆盖糖酵解和TCA循环通路、磷酸戊糖途径、 氨基酸代谢、脂肪酸代谢、 一碳代谢、 核苷酸代谢通路等。丰富的个性化标记定制经验–[U-13C6]-Fructose,[U-13C16]-Palmitate, [U-13C3]-Serine,[U-13C2]-Glycine, [U-13C3]-Alanine, [U-13C3]-Pyruvate, [U-13C4]Malic Acid, [U-13C18]-Oleic Acid, 13CO2, 15N-NH4CL, [1, 2-13C2]-Glucose, [2,3,3-D3]-Serine, [2,3-13C2]Alanine, [1,2,3-13C3]-Choline等。历经数年项目积累,检测各类贴壁细胞、悬浮细胞、菌体、培养液、线粒体、组织、粪便等样本类型,涵盖多发性骨髓瘤、肝癌、线粒体遗传代谢病、免疫细胞活性与疾病、心血管疾病等多个研究方向,合作项目成果突出,文章平均IF >10+。

END
