
文献解读
客户案例 | 多组学分析确定炎症性肠病IBD的微生物与代谢诊断标志物
肠道菌群和代谢物的改变与人类健康和多种疾病有关,包括炎症性肠病(IBD),其作为一组以慢性肠道炎症为特征的疾病,包括克罗恩病(UC)和溃疡性结肠炎(CD),尽管已有研究关注IBD的微生物组和代谢组的变化,但在不同研究之间存在差异,而多组学整合分析在此领域的应用仍相对有限。
2023年11月,上海交通大学医学院附属仁济医院洪洁教授/陈豪燕教授/房静远教授及团队在国际知名期刊Nature Communications上正式发表了题为“Microbiome and metabolome features in inflammatory bowel disease via multi-omics integration analyses across cohorts”的研究性文章,通过整合多组学数据(粪便代谢组和宏基因组)跨队列综合分析(cross-cohort integrative analysis,CCIA),确定了这些队列中肠病相关的肠道微生物群的诊断标记物,创建诊断模型,为这一疾病的精准治疗提供更为全面的认识。(麦特绘谱为本研究提供Q300全定量代谢组检测服务)

研究材料
来自四个不同地区/国家的:
9个宏基因组队列粪便样本(1363例)
4个代谢组学队列粪便样本(398例)
研究技术
(1) 宏基因组
(2) 非靶向代谢组
(3) 靶向全定量代谢组
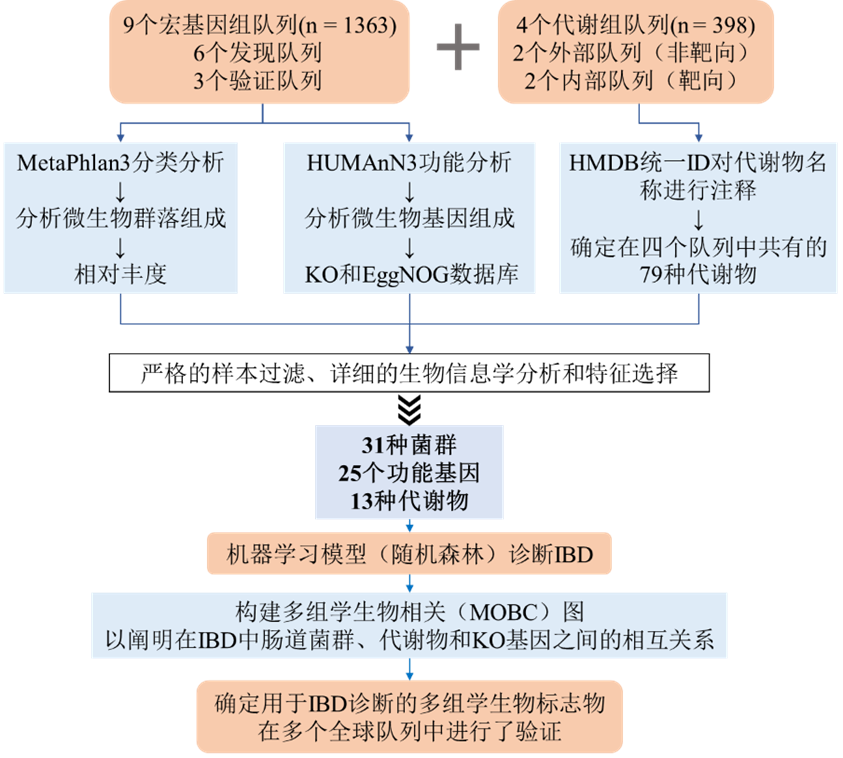
图1. 跨队列整合分析工作流程图
数据集和分析流程
纳入4个不同地区/国家的9个宏基因组和4个代谢组学队列,挖掘IBD相关的微生物诊断标记物。其中代谢组学2个外部队列(USA-NL队列)采用了非靶向代谢组学进行检测,而2个内部队列(CHN队列)则采用了靶向代谢组学进行检测。为了确保生物信息分析的一致性,采用了处理多组学测序数据的bioBakery平台对所有原始测序数据进行重新处理,其中MetaPhlan3工具可以标记基因数据库注释宏基因组数据的物种组成,HUMAnN3则可以准确有效地获得微生物代谢途径和功能模块信息。此外,通过使用Human Metabolome Database(HMDB)统一ID对代谢物名称进行注释,确定了在四个队列中共有的79种代谢物,用于代谢组学数据的跨队列分析。
差异分析确定物种和基因水平上的生物标记物
宏基因组功能分析中,主坐标分析(PCoA)显示两组和队列之间微生物组成存在显著差异,在其中最具差异的74种肠道细菌物种中筛选31种作为后续随机森林建模的特征物种。
使用KEGG orthology数据库将来自宏基因组分析的基因家族注释为KO基因,得到了9,270个KO基因,通过严格的FDR筛选方法确定了正常组和IBD患者之间的存在162个差异表达的KO基因。进一步富集分析,揭示了12个途径可能与肠道菌群和疾病相关(FDR < 0.05)。其中细胞周期-弯曲杆菌、双组分系统、脂多糖生物合成和氨酰-tRNA生物合成显示出上调模式;还观察到多个途径的下调,如丙酸代谢、磷酸转移酶系统(PTS)、苯乙烯降解、糖酵解/糖异生和氨基酸的生物合成。尽管来自不同地区或国家的IBD患者在饮食和遗传学方面存在显著差异,但观察到其肠道微生物群存在的一致改变模式。跨队列综合分析发现IBD患者共生肠菌显著减少,Asaccharobacter celatus和Gemmiger formicilis在6个不同的IBD队列中出现耗竭。
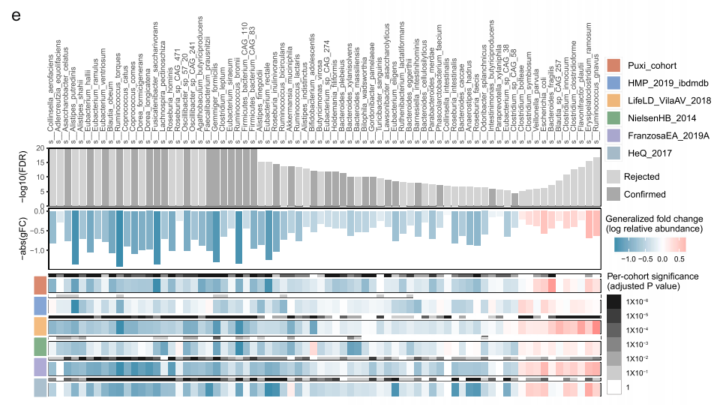
图2. 跨队列鉴定诊断IBD的菌群生物标志物
不同IBD队列中代谢组学改变&利用差异标志代谢物进行IBD诊断
本研究进行了代谢物差异分析并确定了78种代谢物,其中大多数属于三大营养物质,包括氨基酸、碳水化合物和脂肪酸,还观察到与三羧酸循环相关的各种有机酸,如丙酮酸、富马酸、马来酸和α-酮戊二酸在IBD患者中富集,表明肠道微生物群的能量代谢发生异常。
通过交叉队列整合分析(CCIA)方法整合了四个代谢组学研究,使用HMDB ID确定了在所有四个队列中普遍存在的79种代谢物。以FDR<0.0001, VIP score>1为筛选标准确定了32个候选代谢物。进一步采用了迭代特征消除(IFE)技术,缩小到13种差异候选代谢物。
基于筛选的31个特征物种和25个KO基因分别构建机器学习方法(随机森林)来诊断IBD。通过10折交叉验证评估了IBD分类模型的准确性,其中包括每个队列内的评估,以及队列间模型转移的外部验证。该模型在所有全球队列中都显示出强大的IBD检测能力(AUROC值范围从0.92到0.98)。
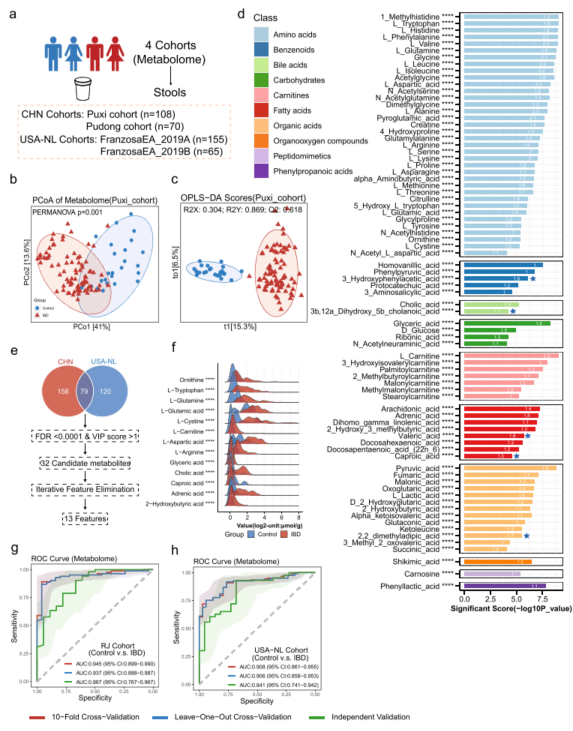
图3. 不同IBD队列中代谢组学改变&利用差异标志代谢物进行IBD诊断
构建多组学生物相关(MOBC)图
通过8个差异性KO基因与代谢和疾病表型两者之间的关联,研究人员构建了多组学生物相关(MOBC)图,鉴定了共生菌群内编码氨酰-tRNA合成酶的四个基因,与KEGG Orthology富集分析中的结果一致,氨酰-tRNA(ARSs)合成途径显著增加,ARSs在蛋白质合成中起着不可或缺的作用,突出了肠道微生物生物转化缺陷和氨酰-tRNA合成酶的显著变化。
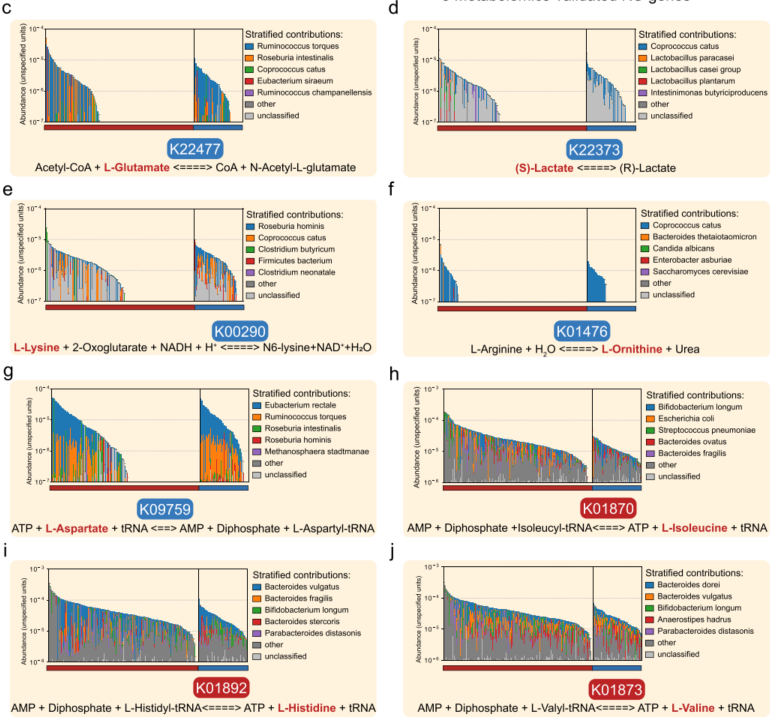
图4. IBD患者肠道微生物群MOBC的构建
小结
本研究中整合了多个队列的宏基因组和代谢组数据,筛选鉴定了31个物种、25个KO基因和13种代谢物,以区分正常对照组和IBD病例。这些生物标志物与目前用于诊断IBD的侵入性金标准——结肠镜检查相比,有力证明了利用肠道粪便微生物群和代谢物作为非侵入性诊断方法的潜力,有助于早期发现和预测IBD,促进及时干预并降低并发症风险。
此外本研究显著提升了用于诊断IBD的机器学习模型性能。从多组学数据中找出了一部分标记物,表现出区分UC和CD的显着能力,平均AUROC值高达0.8。这一发现为非侵入性生物标志物的发展提供了前景,这些标志物可能在对IBD的不同亚型进行临床分类中发挥关键作用。
原文链接
Microbiome and metabolome features in inflammatory bowel disease via multi-omics integration analyses across cohorts. Nature Communications. 2023.
请扫描二维码阅读原文

绘谱帮你测
上述研究在多个队列观察到了微生物组和代谢组的一致性模式,强调了这些关联的鲁棒性,这种独立的队列验证增强了微生物和代谢特征作为IBD生物标志物或治疗靶点的可信度,研究结果值得关注。
麦特绘谱为本研究提供Q300全定量代谢组学分析服务,截至2023年6月,Q300技术已协助客户发表70余篇SCI文章,平均IF>10,成果涵盖肿瘤/癌症、代谢性疾病、免疫疾病、神经系统性疾病、衰老、环境毒理学、中医中药等研究领域。
