
文献解读
绘谱导读 | 抗铁死亡、抗炎、稳神经:菌群代谢物是主角还是NPC?「202401」
导读聚焦
肠道菌代谢物重磅研究:
• 首当其冲——色氨酸明星通路:Cell Host & Microbe发表的一篇研究发现肠道上皮细胞Gpr35通过调节微生物色氨酸代谢信号,经由其菌群代谢物吲哚甲醛(IAld)的神经可塑性发挥抗抑郁作用。来自Nature Cell Biology的论文证实,另一个种色氨酸肠道厌氧菌代谢产物吲哚-3-丙烯酸IDA通过抑制铁死亡促进结直肠癌发展;
• 全新发现——胆汁酸:Gut Microbes一文指出源于肠菌代谢的12-酮胆酸可抑制结肠先天淋巴细胞分泌IL-17A以防止溃疡性结肠炎加重;
• 本期彩蛋——组氨酸:来自Cell Report Medicine的论文报道肠道菌群对组氨酸分解代谢有助于缓解NAFLD,组氨酸通过PI3K-AKT信号通路改善肝脏脂肪代谢。
导读目录
1. Cell Host & Microbe | Gpr35缺失引发抑郁样行为:肠道微生物代谢轴调节
2. Nature Cell Biology | 色氨酸代谢物IDA通过抑制铁死亡促进结直肠癌发展
3. Molecular Cell | 琥珀酰转移酶OXCT1通过琥珀酰化LACTB导致肝细胞癌
4. Nature Communications | 肠上皮细胞中PKCλ/ι缺乏而加强SREBP2驱动的胆固醇生物合成可促进侵袭性锯齿状肿瘤的发生
5. Gut Microbes | 肠道微生物群衍生的12-酮胆酸可抑制结肠第3组先天淋巴细胞分泌IL-17A以防止溃疡性结肠炎急性加重
6. Cell Report Medicine | 由肠道微生物群产生的组氨酸分解代谢对伴有病态肥胖的NAFLD患者具有潜在治疗意义
7. Science Translational MeDicine | 野生型IDH1通过激活丝氨酸生物合成途径维持非小细胞肺癌的干性和化学耐药
8. Nature Communications | Mic19耗竭损害内质网-线粒体互作并引发肝脏疾病
资源领取
本期导读文献原文,请在公众号后台回复“2024年1月绘谱导读”,即可获取资源链接。
一、Cell Host & Microbe | Gpr35缺失引发抑郁样行为:肠道微生物代谢轴调节

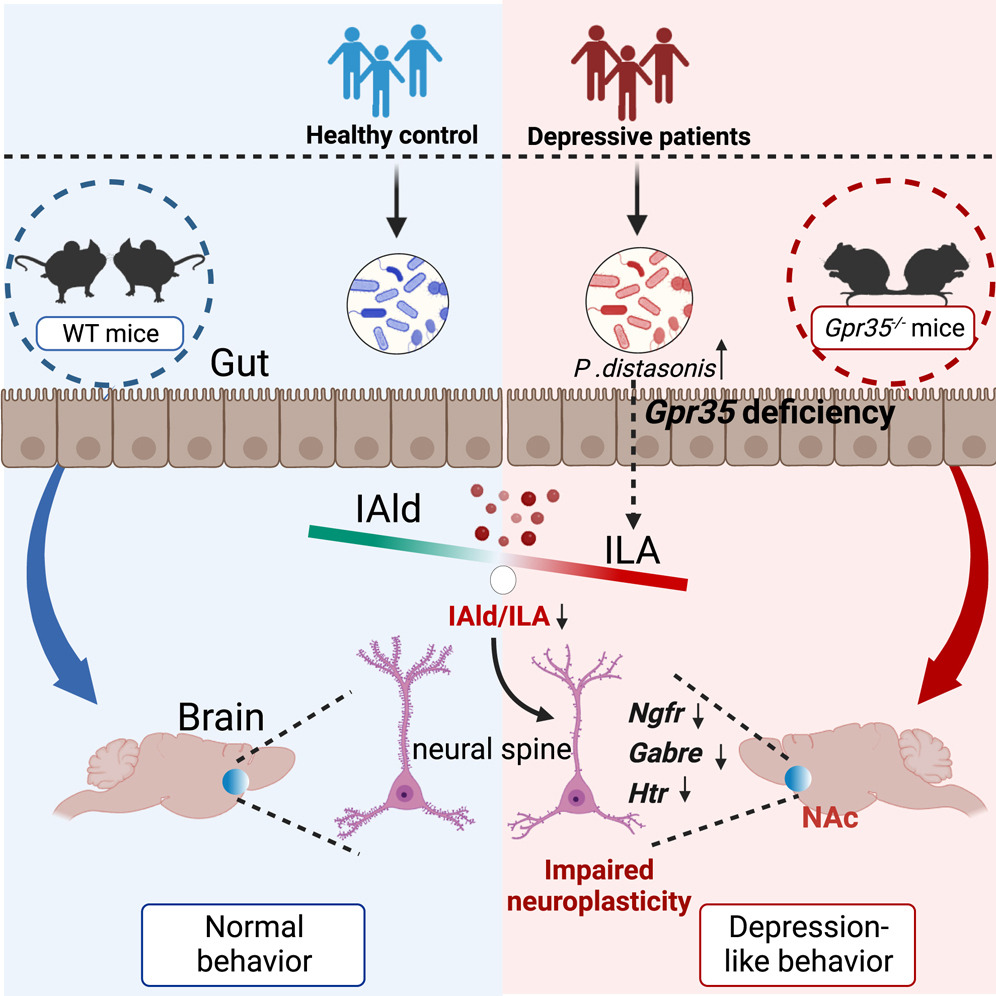
重度抑郁症(MDD)是一种影响全球超过35亿人的神经精神疾病,患者通常伴有躯体症状,特别是胃肠功能障碍,其原因涉及遗传和环境等复杂因素的互作。然而,目前肠道微生态失衡及其通过肠-脑轴的信号传递机制仍不清晰。因此,本研究通过小鼠体内外及临床实验,证实了肠道上皮细胞Gpr35通过调节微生物色氨酸代谢信号在抑郁样行为调控中发挥关键作用,同时发现细菌代谢物吲哚甲醛(IAld)通过恢复伏隔核神经可塑性发挥抗抑郁作用。
●1. 构建Gpr35基因敲除小鼠(Gpr35-/-)模型,通过行为测试评估焦虑抑郁和社会互动行为。结果显示小鼠表现出上述行为,且在共笼实验中得到改善。
●2. 16S rRNA测序结果表明,肠道微生物群结构发生显著变化,特别是菌种Parabacteroides distasonis的丰度增加。
●3. 通过非靶向以及靶向代谢组学分析,研究吲哚代谢物在血清、肠道、大脑区域(如伏隔核)的变化,其中IAld在Gpr35-/-小鼠样本中显著减少,而同时吲哚-3-乳酸(ILA)水平增加。
●4. 通过粪便微生物群移植(FMT)和共笼实验,验证肠道微生物群对行为的影响,发现IAld补充能够缓解小鼠的抑郁症状。
●5. 相关分析结果表明,IAld促进了神经元的生长,而ILA则没有这种效果,且ILA的存在减弱了IAld的促进作用,表明肠道G蛋白偶联受体Gpr35通过微生物代谢途径调节神经可塑性和抑郁症状。
参考文献:
A Gpr35-tuned gut microbe-brain metabolic axis regulates depressive-like behavior. Cell Host & Microbe. 2023.
二、Nature Cell Biology | 色氨酸代谢物IDA通过抑制铁死亡促进结直肠癌发展

既往研究表明肠道菌群失调会引发多种疾病,包括炎症、胃肠道疾病和肿瘤。最新发现肠道微生物通过产生致癌微生物代谢物参与了肿瘤发生的进程,然而其潜在机制目前尚不清楚。本研究发现一种由肠道微生物厌氧菌属产生名为吲哚-3-丙烯酸(3-Indoleacrylic acid ,IDA)的色氨酸代谢物,能够显著促进CRC的进展。IDA通过激活芳香烃受体(AHR)来转录性上调ALDH1A3的表达,该物质利用视黄醇作为底物生成NADH,这对于铁死亡抑制蛋白1(FSP1)介导的还原型辅酶Q10的合成至关重要。
●1. 细胞培养与药物处理结果:在AHR或ALDH1A3基因敲除的细胞中,IDA对铁死亡的抑制作用显著减弱,证实了IDA具有抗铁死亡的特性,IDA介导的肿瘤发展依赖于AHR和ALDH1A3。
●2. 代谢组学分析结果:IDA在厌氧菌属的培养液中被检测到,且IDA的含量与CRC患者的粪便样本中IDA水平正相关。
●3. RNA-seq测序结果:IDA处理后,ALDH1A3基因在HT29细胞中的表达显著上调,提示IDA可能通过AHR途径调控ALDH1A3。
●4. Western blotting结果表明,IDA处理后,ALDH1A3蛋白水平在HT29和HT1080细胞中显著增加,进一步证实IDA对ALDH1A3表达的调控作用。
●5. 相关分析表明:IDA介导的癌细胞铁死亡抵抗,依赖于AHR-ALDH1A3-FSP1信号轴,而阻断IDA-AHR-ALDH1A3-FSP1通路可能有利于结直肠癌的治疗。

参考文献:
Gut microbial metabolite facilitates colorectal cancer development via ferroptosis inhibition. Nature Cell Biology. 2024.
三、Molecular Cell | 琥珀酰转移酶OXCT1通过琥珀酰化LACTB导致肝细胞癌

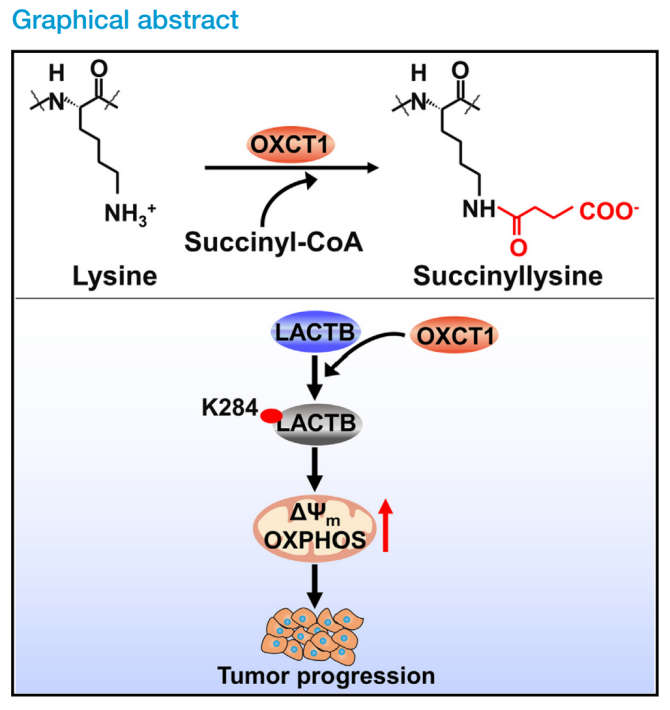
代谢重编程是癌症的一个重要特征,与翻译后蛋白修饰(PTM)密切相关。赖氨酸琥珀酰化是最近发现的一种参与调节蛋白质功能的PTM,但其调节机制及其在肿瘤进展中的作用尚不清楚。前期发现OXCT1在肝细胞癌(HCC)中水平升高激活酮体的分解代谢,从而产生能量并促进癌细胞存活。本研究证实了OXCT1的赖氨酸琥珀酰转移酶功能,并强调了HCC预后与LACTB K284琥珀酰化之间的联系,成为具有潜在开发价值的生物标志物和治疗靶点。
●1. 采用Western blot(WB)检测HCC细胞、HCC病人和小鼠肿瘤组织发现,OXCT1表达升高可促进肝癌细胞和组织中蛋白质赖氨酸琥珀酰化。进一步发现OXCT1对广泛赖氨酸琥珀酰化的影响并不涉及调节细胞内琥珀酰辅酶A水平。
●2. 通过4D定量琥珀酰蛋白质组学分析发现,在OXCT1过表达(OE)细胞中,LACTB蛋白中的几个赖氨酸残基被高度琥珀酰化。并且WB分析显示,LACTB中的残基K284是OXCT1介导的LACTB高琥珀酰化所必需的。
●3. 利用纯化的重组OXCT1和LACTB蛋白进行了体外琥珀酰化实验,并结合WB等实验方法发现,OXCT1通过其结构域V直接与LACTB相互作用,并依靠结构域IV促进琥珀酰基从琥珀酰辅酶A转移到OXCT1,进而转移到LACTB K284残基,最终促进K284的琥珀酰化。
●4. 通过测定细胞生长曲线和分析小鼠肝脏肿瘤等实验发现,OXCT1介导的LACTB K284琥珀酰化消除了LACTB蛋白水解功能,通过提高线粒体PISD蛋白和PE水平,增强了线粒体功能并促进了肿瘤细胞生长。
●5. 在临床HCC病变组织中发现OXCT1和LACTB K284琥珀酰化水平高于邻近非癌组织。并且LACTB K284琥珀酰化可作为HCC患者可靠的预后标志物。
参考文献:
OXCT1 functions as a succinyltransferase, contributing to hepatocellular carcinoma via succinylating LACTB. Mol Cell. 2023.
四、Nature Communications | 肠上皮细胞中PKCλ/ι缺乏而加强SREBP2驱动的胆固醇生物合成可促进侵袭性锯齿状肿瘤的发生

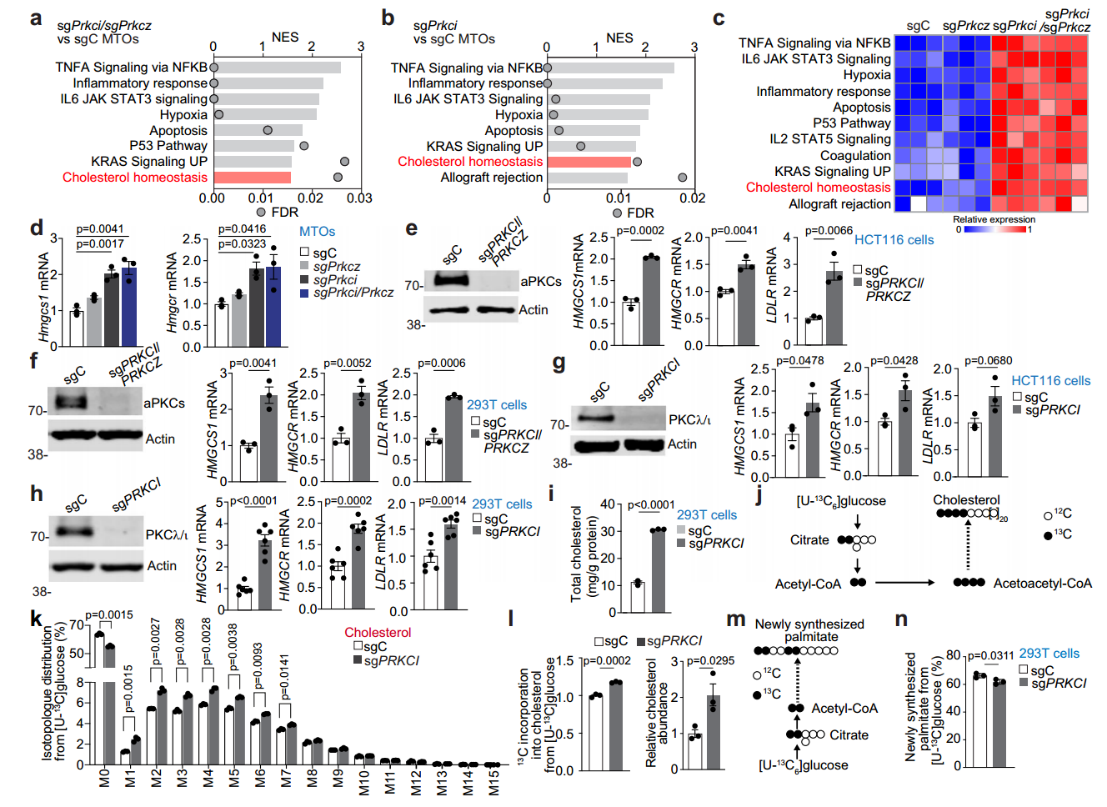
结直肠癌(CRC)起源于两种组织学上可识别的癌前状态:常规腺瘤(CA)和锯齿状途径,其中,通过锯齿状途径调节CRC发生和进展的代谢和信号通路在很大程度上是未知的。本研究发现,低aPKC表达(锯齿状肿瘤的驱动特征)通过抑制其伴侣蛋白SCAP的泛素介导的降解级联反应来上调SREBP2的活性,随后的胆固醇生物合成失调增加了胆固醇的产生,并使肿瘤细胞对这种代谢途径产生依赖,从而产生治疗脆弱性。
●1. 通过转录组学的基因集富集(GSEA)分析发现,小鼠肿瘤和非肿瘤肠道组织表现出胆固醇稳态基因特征的富集。采用体内代谢示踪实验发现,aPKC缺乏会使小鼠肿瘤和非肿瘤肠道组织胆固醇中的生物合成增加。利用scRNA-seq发现胆固醇相关基因特征中表达最高。
●2. 以小鼠肿瘤类器官(MTO)、HCT116细胞和293 T细胞为实验材料,结合转录组学分析,PKCλ/ι缺失和aPKC缺失均使胆固醇升高。并通过13C标记葡萄糖的代谢示踪实验发现,PKCλ/ι缺失使细胞中胆固醇生物合成上调,而脂肪酸合成没有增加。
●3. 通过小鼠肿瘤转录组学发现aPKC缺失和PKCλ/ι缺失均使胆固醇生物合成的主要转录因子SREBP2靶基因的表达上调。并通过对人类锯齿状肿瘤转录组分析也证实胆固醇代谢失调与aPKC低表达相关。
●4. 通过体外磷酸化检测和脉冲追踪实验等一系列实验证明PKCλ/ι介导的磷酸化降低了SCAP的稳定性,PKCλ/ι介导的磷酸化通过TRC8泛素化促进SCAP降解。
●5. 胆固醇生物合成抑制剂阿托伐他汀和双嘧达莫联合使用,不仅对PKCλ/ι缺陷细胞的生长起到抑制作用,还会抑制aPKC缺乏的小鼠肿瘤生长。
参考文献:
Enhanced SREBP2-driven cholesterol biosynthesis by PKCλ/ι deficiency in intestinal epithelial cells promotes aggressive serrated tumorigenesis. Nat Commun. 2023.
五、Gut Microbes | 肠道微生物群衍生的12-酮胆酸可抑制结肠第3组先天淋巴细胞分泌IL-17A以防止溃疡性结肠炎急性加重

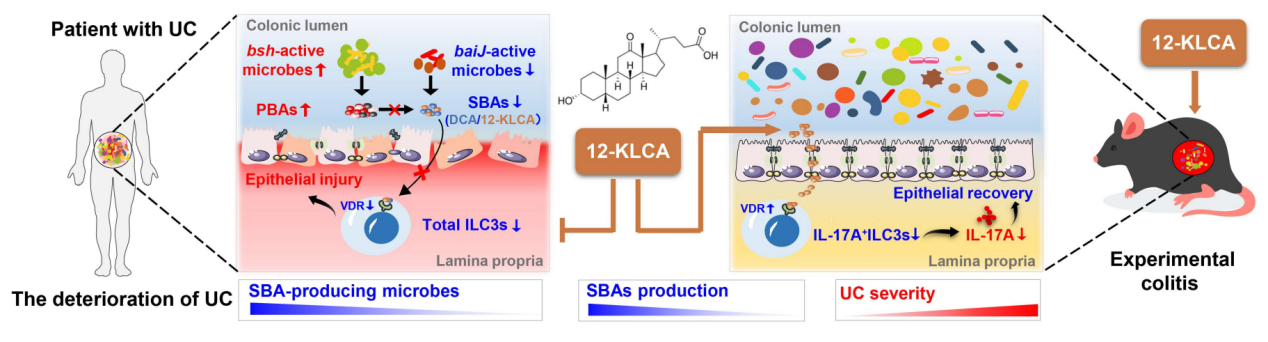
已知肠道菌群失调和代谢紊乱是溃疡性结肠炎(UC)的主要诱因,然而它们在调节对肠道健康至关重要的第3组先天淋巴细胞(ILC3)中的作用在疾病严重程度的发展过程中仍未得到探索。本研究通过肠道菌群测序和靶向胆汁酸代谢发现,12-酮胆酸(12-KLCA)可通过抑制结肠ILC3s中IL-17A的分泌而发挥抗炎作用。
●1. 基于来自不同地区人群粪便样本的16s菌群测序结果表明,重度UC患者的菌群结构不同于轻度和中度UC患者。随后通过LEfSe分析和随机森林鉴定了与UC疾病严重程度相关的指示物种,如Anaerostipeshadrus、乳酸菌及大肠杆菌分别与中度和重度UC相关。
●2. 粪便非靶向代谢组学研究结果表明,胆汁酸生物合成是差异显著代谢物介导的最关键通路。靶向胆汁酸代谢组学研究发现UC患者的肠道次级胆汁酸生物合成缺乏。相关性分析及qPCR检测共同揭示与疾病严重程度相关的菌群与差异次级胆汁酸存在相互作用。
●3. 研究通过菌粪移植实验发现,源自重度UC患者的肠道菌群可诱导受体小鼠产生结肠炎症,小鼠结肠Th17和ILC3s的比例降低,脱氧胆酸和12-酮胆酸大幅度减少。补充12-酮胆酸可有效阻止结肠炎小鼠急性恶化,并促进结肠炎小鼠结肠ILC3s表达核受体VDR而抑制促炎因子IL-17A的分泌。
参考文献:
Gut microbiota-derived 12-ketolithocholic acid suppresses the IL-17A secretion from colonic group 3 innate lymphoid cells to prevent the acute exacerbation of ulcerative colitis. Gut Microbes. 2023.
六、Cell Report Medicine | 由肠道微生物群产生的组氨酸分解代谢对伴有病态肥胖的NAFLD患者具有潜在治疗意义

肠道菌群与非酒精性脂肪性肝病(NAFLD)的病理生理有关。组氨酸是微生物群的关键能量来源,但目前尚不清楚肠道菌群、组氨酸代谢和NAFLD之间是否存在联系。本研究通过人群队列及三种动物模型,借助血浆代谢组学、粪便宏基因组学、肝转录组学和菌粪移植等方法,发现肠道菌群对组氨酸分解代谢有助于缓解NAFLD。
●1. 基于人群队列的血浆代谢组学结果表明,血浆组氨酸水平降低与人体肝脏脂肪变性增加有关。肝脏转录组学结果揭示,胰岛素信号、炎症和微量胺相关受体1(TAAR1)的肝脏转录组特征与血浆组氨酸水平相关。
●2. 进一步探究组氨酸在NAFLD中的作用和潜在机制,发现补充组氨酸可调节人原代肝细胞中的NAFLD,且PI3K-AKT信号转导是与血浆组氨酸水平最相关的通路之一。此外,补充组氨酸可缓解NAFLD模型小鼠、大鼠和果蝇及ob/ob小鼠体内/外肝脂肪变性,减少新发肝脏脂肪生成。
●3. 宏基因组学分析发现变形菌与循环组氨酸水平呈负相关。补充组氨酸可改善NAFLD小鼠的肠道菌群失调特征。
●4. 菌粪移植实验揭示,来自低组氨酸供体的肠道微生物群促进肝脏甘油三酯(TG)积累;携带阴沟肠杆菌的无菌果蝇单定植会增加TG积累和降低组氨酸含量,进而诱导NAFLD样表型。

参考文献:
Potential therapeutic implications of histidine catabolism by the gut microbiota in NAFLD patients with morbid obesity. Cell Report Medicine. 2023.
七、Science Translational MeDicine | 野生型IDH1通过激活丝氨酸生物合成途径维持非小细胞肺癌的干性和化学耐药

肿瘤启动细胞(TICs)被认为是肿瘤发生的驱动力,是复发、远处转移和治耐药性的根源,其表现出代谢可塑性以适应恶劣的微环境和生物合成需求。异柠檬酸脱氢酶1 (IDH1)是一种在代谢重重编程、氧化还原稳态和DNA修复中起作用的关键酶,且IDH1WT(野生型IDH1) 在各种恶性肿瘤中高表达,并与治疗耐药、肿瘤细胞生存优势和患者预后不良相关,但其在NSCLC(非小细胞肺癌)中的作用未知。本研究揭示了IDH1WT在丝氨酸代谢中的作用,及其作为NSCLC中根除TICs和克服吉西他滨化疗抗性的潜在治疗靶点。
●1. 利用Ctrl组和IDH1沉默组的细胞进行非靶向代谢组学分析发现:5-甲基四氢叶酸显著降低,且IDH1沉默显著降低了参与丝氨酸从头合成的两种关键酶PHGDH和PSAT1的表达。免疫组化实验发现,人NSCLC组织中IDH1、PHGDH和PSAT1蛋白丰度显著高于邻近正常样本。
●2. 通过蛋白质组学和免疫共沉淀分析发现,底物结合域2 (SBD2)和C末端调控蛋白(RBD)可以与IDH1共免疫沉淀,进一步采用RNA免疫沉淀法验证了FXR1与PSAT1 mRNA的结合。
●3. 通过13C6标记葡萄糖代谢流分析发现,在IDH1沉默细胞中,除了反映异柠檬酸脱氢酶活性的代谢产物外,13C并入丝氨酸和甘氨酸以及丝氨酸和甘氨酸的总丰度在IDH1 KD细胞中显著降低。
●4. 进一步研究发现,IDH1敲除降低了嘧啶相关代谢物,增强了吉西他滨诱导的PC-9细胞死亡。

参考文献:
Wild-type IDH1 maintains NSCLC stemness and chemoresistance through activation of the serine biosynthetic pathway. Sci Transl Med. 2023.
八、Nature Communications | Mic19耗竭损害内质网-线粒体互作并引发肝脏疾病

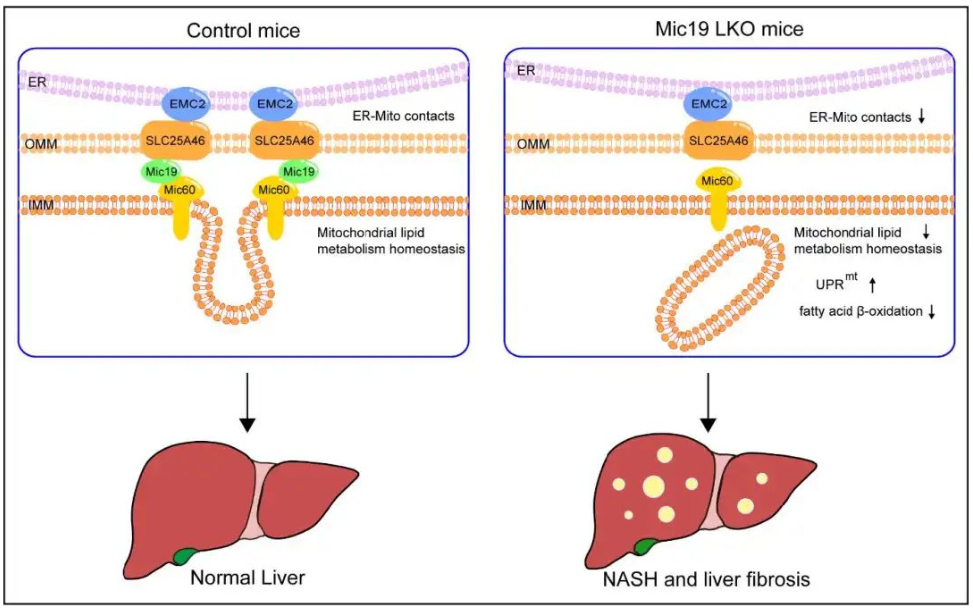
内质网-线粒体互作对脂质运输、合成和代谢的调节至关重要。MICOS复合体是一种多亚基蛋白质复合体,可以稳定线粒体内膜结构和线粒体稳态,Mic19是MICOS配合物的核心亚基之一,影响着MICOS配合物的稳定性。然而,其分子机制和生理功能尚不清楚。本研究明确了EMC2-SLC25A46-Mic19轴是调控内质网-线粒体互作及线粒体脂质代谢新途径,并揭示了内质网-线粒体互作障碍是非酒精性脂肪肝和肝纤维化发生发展的新机制。
●1. 通过免疫共沉淀实验发现,Mic19与线粒体外膜蛋白SLC25A46蛋白结合,SLC25A46与内质网膜蛋白EMC2相互作用。进一步研究发现Mic19和SLC25A46可以相互调节对方的蛋白水平。
●2. 通过脂质组学分析发现,Mic19 LKO小鼠肝脏线粒体部分磷脂酸(PA)、心磷脂(CL)、磷脂酰胆碱(PC)、磷脂酰丝氨酸(PS)和磷脂酰乙醇胺(PE)丰度降低,Mic19的缺失损害了线粒体磷脂的代谢。进一步的脂质组学分析显示,Mic19基因敲除或SLC25A46基因敲低细胞的线粒体分离物中的CL水平降低。
●3. 通过转录组测序发现,在mic19 LKO小鼠肝脏中与脂质代谢相关的基因,尤其是脂肪酸相关代谢的基因下调,禁食条件下Mic19 LKO小鼠肝脏中一些线粒体β-氧化基因Cpt1a、Cpt2、Acad9和Acads的mRNA水平显著降低。
参考文献:
Mic19 depletion impairs endoplasmic reticulum-mitochondrial contacts and mitochondrial lipid metabolism and triggers liver disease. Nature Communications. 2024.
END
