
在线讲堂
公开课回顾 | 肠道菌群和代谢组学数据常用关联分析方法介绍
5月27日绘谱学堂第十一期公开课–《肠道菌群和代谢组学数据常用关联分析方法介绍》由上海交通大学附属第六人民医院转化医学中心贾伟教授课题组陈天璐副研究员主讲。
本期课程首先系统介绍了肠道菌群和代谢组学数据关联分析可以使用的方法,包括方法的原理、相互的关联和方法特点。然后分别介绍两个新的关联方法(工具):多维方法–3MCor,单维方法—GRaMM,这两种方法是陈博以代谢组和微生物组的关联分析而设计研发的,但它们也适用于其他组学之间、组学-表型之间、组学和临床指标之间的相关分析。最后通过两个应用实例来阐述这些关联分析方法如何配合使用从而达到更好的效果。
多组学中,代谢组与微生物组能够很紧密的互作,一些高水平杂志频频报道了这两个组学的联合使用分析。方法上,关联分析是常用的方式。目前,这些方法可以大概分为两个类别:单维分析,即衡量变量和变量之间的关联;多维分析,即计算矩阵和矩阵之间整体先降维,然后用降维后的组合变量来代表整个数据集进行相关分析。很多方法还可以给出具体变量重要性程度。此外,还可以按照线性&非线性、参数&非参数以及是否考虑协变量进行分类。

本课程概述了当前具有代表性的方法,这些在R里面都有对应的函数包,网上也可搜索到相关资源。
单维方法:
(1)Cosine similarity (300BC):只考察向量的方向,不考虑长度;
(2)Pearson correction:要求变量是线性的且服从正态分布;
(3)Spearman rank correction:非参方法,统计效能低于Pearson;
(4)Mutual information(MI):以非线性相关为主,只衡量关联的强弱,不考虑方向;
(5)Maximal information coefficient(MIC, 2011)
多维方法:
(1)Mantel test:简单灵活;
(2)CCA family:降维过程类似PCA;
(3)WGCNA:加权基因共表达网络分析,最初用于基因组或者转录组数据的分析。
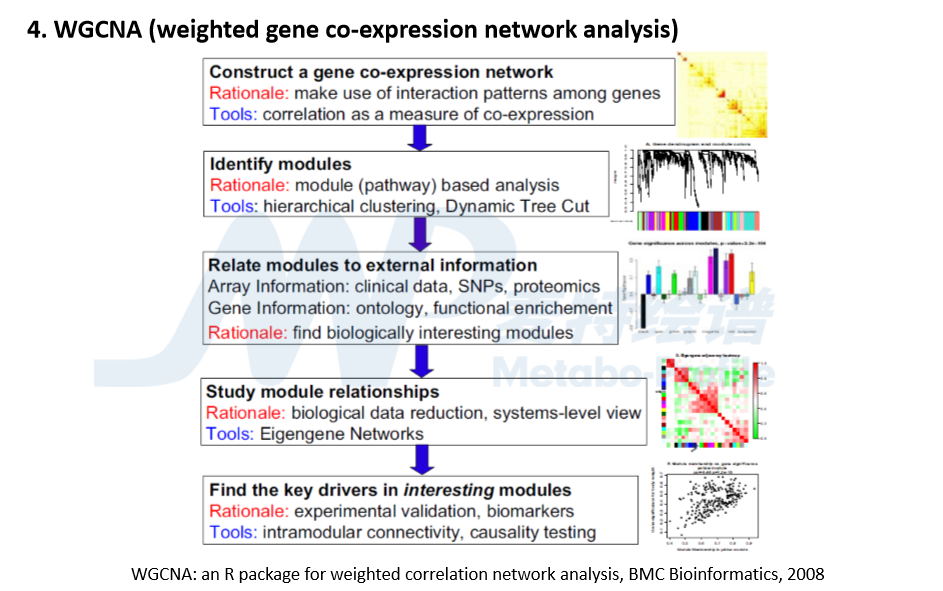
这些方法虽然应用广泛,也有些来自于其他领域,但它们都不是为跨组学分析而设计的,不是为代谢组和微生物组的相关而量身定制的,所以贾伟教授团队根据这些方法的功能和性能做了比较和评估,认识到跨组学存在着几大难点问题:组学数据体量大,变量多,快速找到相关关系很有难度;不同组学特性不同,所以不同的方法会有不同的要求;生物学问题要考虑协变量,不能只考虑两个变量本身;目前多数方法是线性的,或者是单调相关关系的,而非线性的关系还是应该考虑进去。基于前期的借鉴和总结,开发了两个新方法:3MCor和GRaMM,这两个方法基本能够解决上面存在的几个问题。
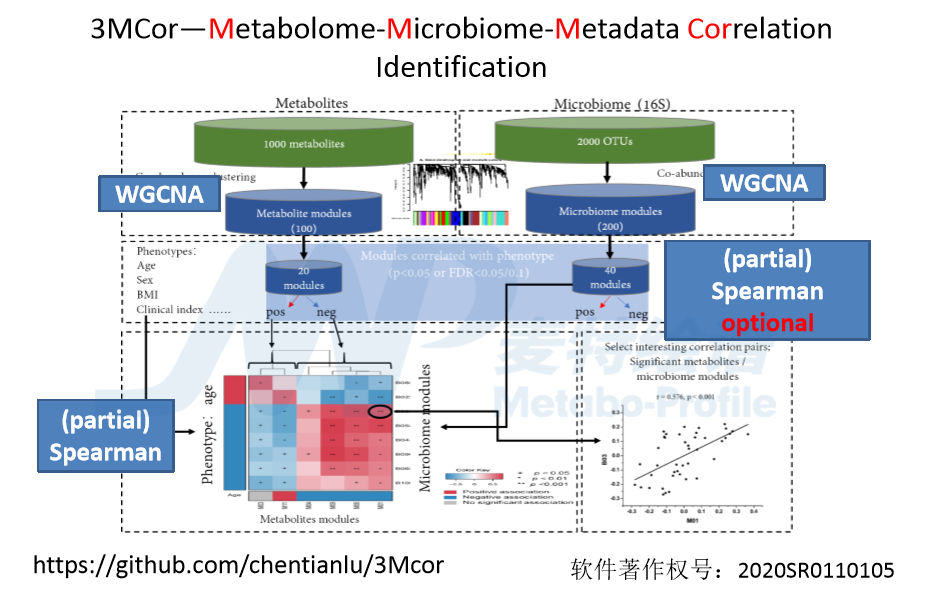
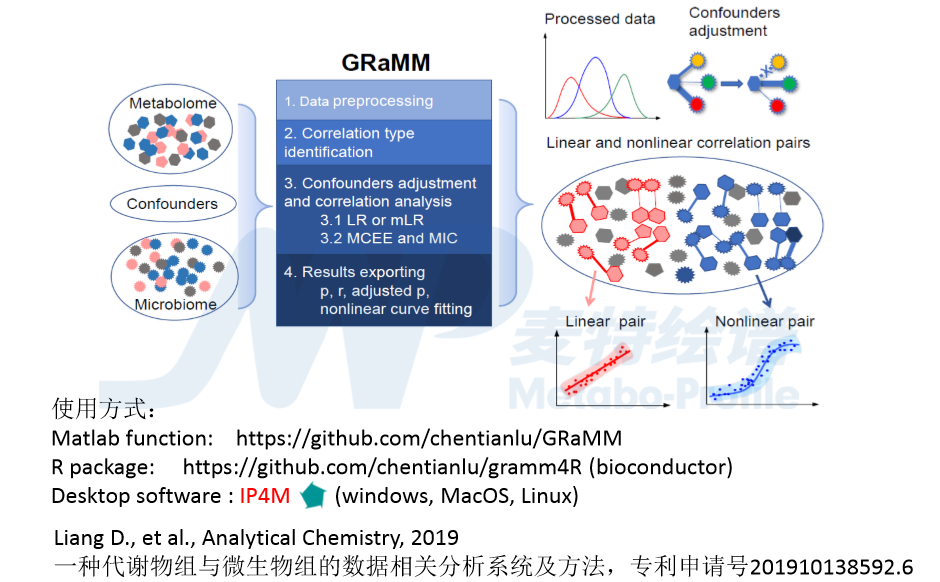
3MCor是一个多维方法,它有两个突出特点:不止考虑了两个组学,还考虑了表型,通过这种方式找到的相关对就会有更加明确地生物学意义;速度快,结果的可重复性比较强。GRaMM是一个单维方法,针对3MCor的不足进行设计的,该方法把Linear Regression、MIC、Centered log-ratio以及MCEE四种方法联合起来进行搭配使用。
相关分析的两个应用实例:
(1)Strategy for association study on intestinal microbiome and brain metabolome across lifespan of rats. Analytical Chemistry. 2018
(2)Age-related compositional changes and correlations of gut microbiome, serum metabolome, and immune factor in rats. GeroScience. 2020

更多代谢组学研究方法欢迎咨询
Q&A
Q1. 文章中的图是否都用R做的?
A1:绝大多数基础的图都可以用R做,但R做的图不能直接用到文章中,后期还需要整合以及调整等。
Q2. 3MCor和GRaMM对样本量是否有要求?
A2:样本量越大越好,但是测试表明,当样本量达到10个,比如代谢组和微生物组分别都达到10,也可以进行分析,只是这种情况下非线性的相关对就没有了。若样本量少于10,就没必要用这种复杂的方法做了。
Q3. 若16S和宏基因组都测了,在分析时如何选择?
A3: 这两种结果会有重叠部分,也会有不同的部分,第2个案例就是做了这两种检测,最后选择两种结果一致的部分进行后续深入的研究。
Q4. 疾病的代谢组和基因组进行关联分析是否有意义,这种分析的样本如果不是来自同一批人群是否可以?
A4:是否有意义取决于实验目的和设计,从数据分析角度是有意义的;这种分析的样本必须是配对的样本才能进行关联分析,否则就没有意义。
Q5. 做双组学分析,R和Python哪个更有优势?
A5:两个都可以用的,它们有很多功能是相通的,具体哪个有优势要根据数据分析以及个人使用的情况来选择。
Q6. WGCNA分析结束后如何过渡到单个变量的分析?
A6:WGCNA是一个降维的方法,比较复杂,我们的方法中得到的module就是单变量,即把多个变量变成一个单变量,然后就可以直接用单变量的分析方法。
Q7. 对于代谢组和微生物组能否通过关联分析,分析出具体哪种菌影响哪种代谢产物或者哪种代谢产物影响哪种菌?
A7:相关分析说明不了菌群和代谢物的因果关系,而是起着提示作用,基于此可以先提出假说,后续设计实验进行验证。谁是因谁是果需要通过验证实验来确定,比如添加或剥夺代谢物观察对菌群有何影响,同理,也可以添加菌群或抗生素看对代谢物的影响,不管哪种方式(具体可以参考文献)都需要验证某个因素对疾病发生发展的影响作用。
Q8. 脂质组和蛋白组的关联有没有推荐的做法?
A8:3MCor和GRaMM这两个方法是可以做其他组学的分析,这两个方法都可以尝试。
Q9. 若有14例样本做了粪便宏基因组和菌群宿主共代谢物的检测,可以直接用Spearman关联分析吗?
A9:做是可以做,但数据量比较大,用Spearman分析后会出现强相关的特别多,真实的结果就会被大量其他结果掩盖,效果如何不好考量。但若有明确关注的某个代谢物和某个菌的关联,想做验证的话,用Spearman是没问题的。
Q10. 粪便测的菌群,可否与血清的代谢组做关联分析?
A10:从数据分析的角度可以做。但肠道菌群和代谢组学检测的样本推荐来源于同一个体的相同样本类型并确保生物学重复数及同批次收集,例如同一个体的粪便或肠道内容物数据;算法上对样本类型无特定要求,确保样本类型一致性得到的关联分析可以排除样本类型不同造成的误差而使结果更直观且便于解释。同一个个体的血浆(或血清)代谢数据和粪便肠道菌群数据也可以进行相关性分析,只是两者之间的相关性不如相同样本类型更直观。
Q11. 研究肠道菌和肿瘤代谢组的关联,代谢组样本选择肿瘤组织、血浆还是粪便好?
A11:不同样本类型反映的信息不同,它们是不能相互替代的,需要根据实验目的或者实验预期进行选择,建议都做是最好的。
Q12. 进行菌群和代谢产物检测,取ASD患者的粪便时是否要在同一处取,若选取了多处收集,是否可以用于检测?
A12:一般是在同一处取,然后分成多份,分别进行菌群和代谢物检测;已取多处的话,建议将这些样本混合后再分成多份进行检测分析。

